Alpha-Galactosidase A (GLA) Antibody
195€ (20 µl)
Por favor contáctenos para obtener información detallada sobre el precio y disponibilidad.
Name
Alpha-Galactosidase A (GLA) Antibody
Category
Primary Antibodies
Provider
Abbexa
Reference
abx001425
Tested Applications
ELISA, WB, IF/ICC
Description
GLA Antibody is a Rabbit Polyclonal antibody against GLA. This gene encodes a homodimeric glycoprotein that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. This enzyme predominantly hydrolyzes ceramide trihexoside, and it can catalyze the hydrolysis of melibiose into galactose and glucose. A variety of mutations in this gene affect the synthesis, processing, and stability of this enzyme, which causes Fabry disease, a rare lysosomal storage disorder that results from a failure to catabolize alpha-D-galactosyl glycolipid moieties.
Documentos del producto
Instrucciones
Data sheet
Especificaciones del producto
| Category | Primary Antibodies |
| Immunogen Target | Target: Alpha-Galactosidase A (GLA) Immunogen: Recombinant protein corresponding to GLA. The exact sequence is proprietary. |
| Host | Rabbit |
| Reactivity | Human, Mouse |
| Assay Type | Concentration: > 0.2 mg/ml |
| Recommended Dilution | ELISA: 1 µg/ml, WB: 1/500 - 1/2000, IF/ICC: 1/50 - 1/100. Optimal dilutions/concentrations should be determined by the end user. |
| Clonality | Polyclonal |
| Conjugation | Unconjugated |
| Isotype | IgG |
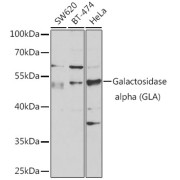
| Observed MW | Calculated MW: 49 kDa Observed MW: 49 kDa |
| Purification | Purified by affinity chromatography. |
| Size 1 | 20 µl |
| Size 2 | 100 µl |
| Size 3 | 2 × 100 µl |
| Form | Liquid |
| Tested Applications | ELISA, WB, IF/ICC |
| Buffer | PBS, pH 7.3, containing 0.02% sodium azide, 50% glycerol. |
| Availability | Shipped within 5-10 working days. |
| Storage | Aliquot and store at -20°C. Avoid repeated freeze/thaw cycles. |
| Dry Ice | No |
| UniProt ID | P06280 |
| Gene ID | 2717 |
| NCBI Accession | NP_000160.1 |
| Background | Antibody anti-GLA |
| Status | RUO |
| Note | THIS PRODUCT IS FOR RESEARCH USE ONLY. NOT FOR USE IN DIAGNOSTIC, THERAPEUTIC OR COSMETIC PROCEDURES. NOT FOR HUMAN OR ANIMAL CONSUMPTION. |
Productos relacionados
Alpha-Galactosidase A (GLA) Antibody
GLA Antibody is a Rabbit Polyclonal antibody against GLA. This gene encodes a homodimeric glycoprotein that hydrolyses t…
Ver producto
Alpha-Galactosidase A (GLA) Antibody
GLA is a homodimeric glycoprotein that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoprote…
Ver producto
Human Alpha-Galactosidase A (GLA) Protein
Human alpha Galactosidase is a recombinant Human protein expressed in E. coli.…
Ver producto